
Россия, Вольск


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 13.08.2021 13:31

КРАЛЯ ИРИНА ИВАНОВНА
Преподаватель общеобразовательных дисциплин
52 года
Местоположение
Специализация
Лекционный материал по ОУД Химия. Лекция 5 4.6. Ионные связи и поляризация
Категория:
Химия
30.07.2021 12:00

Просмотр содержимого документа
«Лекционный материал по ОУД Химия. Лекция 5 4.6. Ионные связи и поляризация»
Лекция 5
4.6. Ионные связи и поляризация
Химические связи с большой долей ионности часто представляют как чисто ионные, и в этих случаях для расчетов характеристик связи и вещества (и прежде всего энергии) используют закон Кулона. При этом ионная связь оказывается ненаправленной и ненасыщаемой, т.е. количество соседних атомов (координационное число) определяется их размерами и соотношением зарядов.
Например, в кристаллах NaCl координационные числа Na+ и Cl одинаковы (6), а в SrF2 различаются в 2 раза (8 и 4). Полярность и ковалентность химической связи определяются дипольным моментом связи, эффективным зарядом атомов, которые связаны с их электроотрицательностями (см. раздел 3.2.2). Некоторая доля ковалентности связи учитывается введением представления о поляризации (деформации) ионов.
Поляризация ионов. Так как электронные плотности атомов и ионов простираются на значительные расстояния от ядер и имеют заметные значения за пределами их эффективных радиусов, то при образовании любой химической связи обязательно имеет место перекрывание облаков, то есть связь не может быть чисто ионной.
Каждый ион можно характеризовать его поляризующим действием, которое зависит от напряженности создаваемого им электрического поля, пропорционального отношению заряда к радиусу (q / r ионный потенциал), и поляризуемостью, то есть способностью их к деформации _ (измеряется в единицах объема х1024 см3). Качественные правила, характеризующие поляризацию ионов, сформулированы К. Фаянсом в 1923 г.
1. Поляризация тем больше, чем меньше размер катиона и больше его заряд, то есть чем больше ионный потенциал (![]() ). Если заряд измерять в единицах заряда ē (1, 2, 3 и т.д.), а r в нанометрах (10-9 м), то ионные потенциалы ряда катионов будут следующими:
). Если заряд измерять в единицах заряда ē (1, 2, 3 и т.д.), а r в нанометрах (10-9 м), то ионные потенциалы ряда катионов будут следующими:
| Li+ | 17 | Be2+ | 64 | B3+ | 150 |
| Na+ | 10 | Mg2+ | 31 | Al3+ | 60 |
| K+ | 8 | Ca2+ | 20 | Ga3+ | 48 |
Поляризующая способность сильно растет с зарядом. Поэтому катионы с большим зарядом (B3+, Si 4+, P5+) образуют только ковалентные соединения. Видно, что по ионному потенциалу элементы более близки в диагональном направлении периодической таблицы: Li+ и Mg2+, Be2+ и Al3+ и т.д.
2. Поляризация тем больше, чем больше отрицательный заряд аниона и больше его радиус; многозарядные анионы по этой причине образуют только ковалентные соединения (P3, As3).
3. Поляризация катионов больше, если у них внешние электроны не имеют конфигурации благородного газа (s2р6), а имеют конфигурацию (n 1)d x ns0 (где х от 1 до 10).
Например, ионы Hg2+ и Ca2+ имеют близкие радиусы (0,116 и 0,114, соответственно) и ионные потенциалы, однако соединения Hg2+ сильно ковалентны, а Ca2+ ионны, что связано с большей поляризацией ионов в соединениях Hg2+.
Поляризация ионов, связанная со степенью ковалентности (К) или ионности (i), сильно влияет на такие свойства веществ как температуры плавления и кипения, растворимость, цветность. Так, увеличение поляризации ионов в ионных кристаллах приводит к снижению температур плавления, например, температуры плавления (C) BeCl2 (450), CaCl2 (772), HgCl2 (276), NaBr (755), MgBr2 (700), AlBr3 (97,5), LiF (870), LiCl (613), LiBr (547), LiI (446).
Растворимость в полярных растворителях уменьшается с увеличением поляризации. Например, в ряду AgF, AgCl, AgBr, AgI поляризация растет из-за увеличения радиуса аниона, а растворимость падает.
2
М
 О 1
О 1

 С О
С О
М О
Н2СО3 и Н2СS3 неустойчивы уже при комнатных температурах, так как катион Н+ имеет очень малый размер ( 10-5 нм) и очень большой ионный потенциал. Температуры разложения других карбонатов также подчиняются этой закономерности (в С): BeCO3 (100), MgCO3 (400), CaCO3 (900).
Изменения в спектрах соединений (цвет) также зависят от поляризации ионов и степени ковалентности, так как их увеличение фактически означает частичный перенос электронной плотности с аниона на катион, что уменьшает энергию, необходимую для переноса действием света. Например, CaI2 бесцветен, так как бесцветны свободные ионы Са2+ и I- , а поляризация в этом соединении слаба. Это значит, что требуется большая энергия квантов света для переноса электрона. В то же время HgI2 и PbI2 окрашены (красный и желтый, соответственно) , что связано с большей поляризующей способностью катионов Hg2+ и Pb2+ и уменьшением энергии для переноса электронов.
4.7. Теории металлической связи
Вещества с металлическими связями металлы обладают рядом особых свойств. К ним относятся высокие тепло- и электропроводность, сплошной спектр поглощения света, а также высокая пластичность многих металлов и образование между ними соединений, не отвечающих валентностям. Эти свойства говорят о том, что металлическая связь делокализована, имеет множество близко расположенных электронных состояний и ненаправленна. Некоторые из этих свойств были описаны моделью свободных электронов и методом МО.
Модель свободных электронов. В этой модели металл представляется как совокупность катионов, образующих остов, как бы погруженных в электронную жидкость, частицы которой (электроны) свободно перемещаются между катионами. Энергия связи определяется как кулоновское взаимодействие между катионами и электронами. Эта теория хорошо описывает свойства щелочных металлов. В частности, структура металлов определяется тем, что минимум энергии системы достигается при плотнейшей упаковке катионов, что имеет место при координационных числах 8 и 12; при этом связь оказывается ненаправленной, так как изменения в геометрии расположения атомов и даже изменения координационного числа в указанных пределах мало изменяют энергию связей. Эта модель, однако, плохо объясняет свойства других металлов и спектры металлов.
Теория молекулярных орбиталей (МО) для металлов. Теория МО для металлов объясняет все их свойства. В этой теории кристалл металла рассматривается как гигантская молекула из N атомов, в которой все атомы взаимодействуют друг с другом (а не только соседние). В этом случае МО будут охватывать весь кристалл. Сами МО образуются путем линейной комбинации АО всех N атомов:
![]() .
.
При этом из N АО образуются N МО. Расчетным и экспериментальным путем (по спектрам) показано, что разница в энергиях (Е) между самой нижней и самой высокой составляет величину порядка обычной химической связи (несколько сотен кДж/моль). Тогда расстояние между соседними МО будет очень малой величиной порядка 1018 Дж/моль (![]() ) (рис. 3.26).
) (рис. 3.26).
Если взять, например, N атомов Li, имеющих по одному валентному электрону на одной атомной орбитали (2s1), то при их взаимодействии образуется столько же МО. Так как на каждой МО может быть 2ē, то лишь половина МО будет занята. Расстояние между соседними МО Е чрезвычайно мало, поэтому поглощение любого кванта энергии (даже тепловой или энергии внешнего поля) вызывает возбуждение электрона; это объясняет сплошной спектр и высокую подвижность электронов.
В случае металлов с полностью заполненными подуровнями (Ве: 2s22р0) имеет место перекрывание зон 2s-и 2р-МО и картина, показанная на рис 3.26, сохраняет свой вид (близкое расположение пустых и заполненных мест), так как обычно соседние пустая и заполненная зоны перекрываются (2s2р).
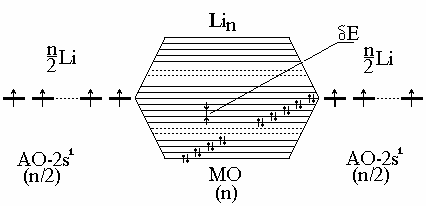
Рис. 4.26. Образование почти сплошной зоны МО в металлах
В случае металлов с незаполненными d-подоболочками dd-взаимодействие приводит к образованию локализованных и направленных ковалентных МО. Этим объясняются высокая твердость и высокие температуры плавления таких металлов. Аналогичная ситуация и в случае f-металлов.
Таким образом, молекулярные орбитали металла образуют почти непрерывную зону разрешенных энергий (этим, в частности, объясняется непрерывный, а не линейчатый, как у атомов, или полосатый, как у молекул, спектр поглощения металлов). Разность между верхней и нижней энергиями (Е) зоны называется шириной зоны. Зона, заполненная электронами, называется валентной. Зона, свободная от электронов и находящаяся выше валентной зоны, зона проводимости. Они могут либо перекрываться, либо не перекрываться друг с другом. Если эти зоны не перекрываются, то между ними существует запрещенная зона с шириной Е.
Ширина запрещенной зоны определяет тип кристалла: металл, полупроводник или диэлектрик (рис. 4.27).
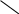

Е 
 Е
Е
Е
 а б в
а б в
Рис. 4.27. Зонная структура металлов (а), полупроводников (б)
и диэлектриков (в)
(верхняя зона – зона проводимости, нижняя – валентная зона)
Теория, с помощью которой объясняют свойства кристаллов, получила название зонной теории. При ширине запрещенной зоны ниже
4 эВ кристаллические вещества проявляют полупроводниковые свойства. При поглощении энергии электроны валентной зоны возбуждаются и переходят в зону проводимости, а в валентной зоне появляются вакансии электронов, которые имеют эффективный положительный заряд; их называют дырками. Наличие подвижных электронов и дырок обеспечивает собственную проводимость полупроводников. Собственную проводимость имеют, например, кремний и германий.
У диэлектриков ширина запрещенной зоны превышает 4 эВ. Для возбуждения электронов требуется очень значительная энергия, нагреванием такого возбуждения достичь невозможно, так как при этом кристалл либо расплавится, либо разрушится.
К диэлектрикам относятся многие вещества с ионными и молекулярными кристаллами, а также некоторые вещества с ковалентными кристаллами, например алмаз (Е = 5,1 эВ) и кварц (Е = 5,2 эВ).
4.8. Кристаллические решетки
Если вещество образует молекулы, полярные или неполярные, то его кристаллы обычно построены из молекул, т.е. имеют молекулярную решетку. Силы, действующие между молекулами, сравнительно слабые. Поэтому вещества с молекулярной решеткой имеют малую твердость, низкие температуры плавления, плохую растворимость в воде. При обычных условиях это, как правило, газы или жидкости.
Среди простых веществ их всего 9: Н2, N2, О2, F2, Cl2, Br2, I2, Р4 и S8.
Среди сложных веществ это большинство органических веществ, которые находятся при обычных условиях в твердом состоянии.
Из неорганических соединений это твердый СО2 (сухой лед), вода (лед), твердые галогеноводороды и многие другие.
Для молекулярных веществ характерна следующая закономерность: вещества с большей молекулярной массой имеют более высокие температуры плавления и кипения.
Вещества с ионным типом связи часто образуют ионные решетки. Это, как правило, ионные проводники, растворяющиеся в полярных растворителях. Они тугоплавки, малолетучи, сравнительно прочны.
Если вещество не является ионным, но и не образует молекул, все связи оказываются одинаково прочными. Образуется атомная кристаллическая решетка. Вещества с атомной решеткой имеют высокие температуры плавления, высокую прочность и твердость. Они практически нерастворимы в воде и других жидкостях. Атомная решетка характерна для бора, углерода, кремния, германия, некоторых соединений этих элементов с другими (BN, SiO2 и др.).
4.9. Водородные связи
Давно было замечено, что простейшие соединения водорода с легкими электроотрицательными элементами, например фтором или кислородом, отличаются от аналогичных соединений с тяжелыми элементами аномально высокими температурами кипения и плавления. На рисунке 3.28 приведены кривые зависимости температур кипения простых водородных соединений р-элементов VVII групп от номера периода.
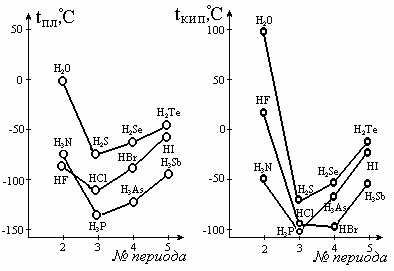
Рис. 4.28. Зависимость температуры плавления (а) и кипения (б)
водородных соединений р-элементов V, VI и VII групп от № периода
Видно, что для них наблюдается последовательное повышение температуры кипения с ростом номера периода, но из нее заметно выпадают вода, фтороводород и аммиак. Экстраполяция температуры на графике дает tкип воды около 80оС. То же происходит с теплотой плавления и испарения. Объяснение этим и другим фактам было дано русскими химиками Ильинским и Бекетовым в 80-х годах XIX века. Они предположили, что между молекулами таких соединений существует особая, водородная, связь. Ее образование объясняется особыми свойствами атома водорода: катион Н+, в отличие от других катионов, является элементарной частицей с резко отличающимися размерами (меньше обычных атомов в 105 раз) и высокой подвижностью.
Поэтому он может осуществлять связь между соседними атомами, если они несут на себе отрицательные заряды. Характер связи при этом близок к ионному. А так как Rэ- Rн+![]() , то угол связи ЭНЭ равен 1800.
, то угол связи ЭНЭ равен 1800.
+ + +
H FH FH F
Благодаря водородным связям молекулы объединяются в димеры и более сложные ассоциаты. Последние могут иметь линейное, разветвленное или кольцевое строение. Ассоциация приводит к повышению температуры кипения, температуры плавления и теплоты парообразования, изменению растворяющей способности и т.д. Энергия водородной связи меньше, чем обычных ковалентных и ионных связей (обычно менее 40 кДж/моль). Она тем больше, чем больше электроотрицательность элемента, Так, энергия водородной связи Н...F составляет около 40, связи Н....О 20, Н...N 8 кДж/моль. Обозначается такая связь обычно пунктиром.
Все рассмотренные примеры относятся к межмолекулярным водородным связям. Нередко водородные связи объединяют части одной и той же молекулы, то есть являются внутримолекулярными. Это характерно для многих органических веществ.
Следует отметить, что внутримолекулярные водородные связи, в отличие от рассмотренных межмолекулярных, понижают температуры и теплоты кипения и плавления веществ. Так, например, орто-нитрофенол, имеющий внутримолекулярную водородную связь, плавится при 450С, а мета-нитрофенол, молекулы которого ассоциированы за счет межмолекулярной водородной связи, при 970С.
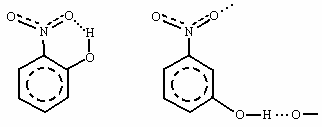
орто-нитрофенол мета-нитрофенол
Водородные связи имеют большое значение в живой и неживой природе. Например, построение белковых молекул, особые свойства воды, формируемые с участием водородных связей, обеспечивают само существование жизни на Земле.
4.10. Межмолекулярные силы Ван-дер-Ваальса
Так как вещества, состоящие из полностью насыщенных электронами молекул (с замкнутыми 8- и 18-электронными оболочками всех атомов, например CH4, N2O5) и не содержащие ионов (H2, Ar, N2), при понижении температуры сжижаются и кристаллизуются, то очевидно, что между молекулами существуют какие-то нерассмотренные выше межмолекулярные взаимодействия. Эти силы были названы именем голландского физика Ван-дер-Ваальса, который внес большой вклад в изучение процессов конденсации газов. Было установлено, что эти силы имеют 3 составляющие.
Ориентационное взаимодействие. Оно проявляется, если вещество состоит из полярных молекул диполей (дипольдипольное взаимодействие). В результате беспорядочного теплового движения молекул при их сближении друг с другом диполи в веществе ориентируются с выигрышем энергии (рис. 3.29). Чем более полярны молекулы, тем сильнее они притягиваются и тем сильнее ориентационное взаимодействие. Такое взаимодействие характерно для полярных молекул (NH3, H2O и др.).
Повышение температуры ослабляет это взаимодействие, так как тепловое движение нарушает взаимную ориентацию молекул. Притяжение полярных молекул быстро уменьшается с расстоянием между ними.
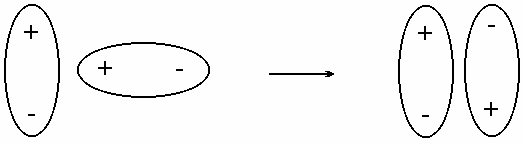
Рис. 4.29. Ориентация диполей
Индукционное взаимодействие. Оно осуществляется, в частности, между полярной и неполярной молекулой и обусловлено тем, что дипольные молекулы индуцируют в соседних молекулах диполи. Индуцированные диполи взаимодействуют между собой и другими диполями, что дает выигрыш в энергии. Энергия этого взаимодействия тем больше, чем больше поляризуемость молекул. Например, в H2S это взаимодействие больше чем в Н2О, так как поляризуемость S2 больше чем O2.
Дисперсионное взаимодействие. Это взаимодействие обусловлено тем, что каждый атом является диполем в любой момент времени, т.к. электрон и ядро являются противоположно заряженными частицами (мгновенный диполь). Если имеется несколько атомов поблизости, то их диполи ориентируются в пространстве ("+" к "") с выигрышем энергии. Такое взаимодействие характерно для любых атомов и молекул. Оно тем больше, чем более тяжелыми являются частицы (больше зарядов больше взаимодействий). Дисперсионное взаимодействие наиболее универсальное, то есть проявляется в любых случаях.
Суммарная энергия межмолекулярного взаимодействия обычно около 10 кДж/моль. Наименьший вклад обычно вносит индукционное взаимодействие. В случае атомов (He, Ar и др.) и неполярных молекул (H2, N2 и др.) наибольший вклад дает дисперсионное взаимодействие.
Все три вида взаимодействия возрастают с ростом молекулярной массы.
По сравнению с ковалентной связью ван-дер-ваальсово взаимодействие очень слабое. Так, если энергия, необходимая для диссоциации молекулы Cl2 на атомы составляет 243 кДж/моль, то энергия сублимации (возгонки) кристаллов Cl2 составляет всего 25 кДж/моль.
4.11. Комплексные соединения.
Определения, составные части и классификация
Комплексными называются соединения, которые можно рассматривать как образованные из более простых:
3KCN + Fe(CN)3 = K3[Fe(CN)6] ,
4NH3 + CuSO4 = [Cu(NH3)4]SO4 .
В квадратные скобки ставят собственно комплекс. Внутри скобок внутренняя координационная сфера, а за скобками внешняя.
Комплексы (комплексные, координационные соединения) это молекулы или ионы, обладающие высокой симметрией, имеющие атом в центре симметрии центральный атом, комплексообразователь, электронная валентность которого больше стехиометрической. Вокруг центрального атома располагаются атомы или группы атомов лиганды, которые и образуют симметричную фигуру.
Например, [Pt(NH3)2Cl2] квадрат с атомом Pt в центре (Vстх. = 2, Vē = 4) и лигандами Cl и NH3; [CoF6]3 октаэдр с атомом Co в центре и лигандами F (Vстх = 3, Vē = 6).


 3
3
F F F
F F

 Cl NH3 \
Cl NH3 \
Pt Со
H3N Cl / \
F F F
В качестве лигандов выступают обычно анионы (F, Cl, CO32), нейтральные молекулы (H2O, NH3) и, очень редко, катионы (например, NO+).
Лиганды могут занимать одно, два, три и более мест вокруг центрального атома. Число мест, занимаемых лигандом, называется дентатностью. Например, все приведенные выше лиганды занимают одно координационное место они монодентатны. У этилендиамина (NH2CH2CH2NH2) два координационных места (у атомов N), он бидентатен; анион этилендиаминтетрауксусной кислоты (ЭДТА) гексадентатен и т.д.
Число координационных мест вокруг центрального атома называется его координационным числом (4 и 6 в приведенных примерах).
В качестве центрального атома в комплексах обычно выступают катионы, иногда атомы и редко отрицательные ионы. Например, в приведенных выше комплексах это катионы Fe2+, Cu2+, Pt2+ и Co3+, в комплексе [Ni(CО)4]0 нейтральный атом Ni; в комплексе H[Co(CO)4] отрицательный ион Co.
Комплексные соединения классифицируются по составу и заряду комплексов:
катионные [Ni(NH3)4]2+, анионные [Co(CN)6 ]3, нейтральные [Co(NH3)4Сl2]0;
по кислотно-основным свойствам: кислоты H[AuCl4]; основания [Ag(NH3)2]OH; соли [Ni(NH3)6]SO4;
по типу (составу) лигандов: гидроксокомплексы K2[Zn(OH)4]; аквакомплексы [Fe(H2O)6]Cl3; ацидокомплексы (лиганды анионы кислот) K4[Fe(CN)6]; комплексы смешанного типа K[Co(NH3)2Cl4], [Pt(NH3)4(H2O)2]Cl4.
Пример 6. Определите заряд комплексного иона, координационное число (к.ч.) и степень окисления комплексообразователя в соединениях:
а) K4[Fe(CN)6]; б) K2[MoF8]; в) [Cr(H2O)2(NH3)4]Cl3.
Решение. Координационное число комплексообразователя равно числу связей c лигандами, координированных вокруг него. Степень окисления комплексообразователя определяется так же, как степень окисления атома в любом соединении, исходя из того, что сумма степеней окисления всех атомов в молекуле равна нулю. Заряды нейтральных молекул (H2O, NH3) равны нулю. Заряды кислотных остатков определяют из формул соответствующих кислот. Заряд комплексного иона равен заряду внешней сферы, но противоположен по знаку. Отсюда:
| Соединение | Заряд комп- лексн. иона | к.ч. центр. атома | Степ. окисл. центр. атома |
| а) K4[Fe(CN)6] | 4 | 6 | +2 |
| б) K2[MoF8] | 2 | 8 | +6 |
| в) [Cr(H2O)2(NH3)4]Cl3 | +3 | 6 | +3 |
Названия комплексов строятся по общим правилам IUPAC : справа налево, лиганды с окончанием -о, анионы с окончанием -ат. Лишь некоторые молекулы-лиганды имеют особые названия, например Н2О и NH3 называют аква и аммин, соответственно.
Пример 7. Дать названия следующим комплексным соединениям:K3[Fe(CN)6], [Cu(NH3)6]SO4, [CoF3(H2O)3].
Решение. K3+[Fe+3(CN)6]3 комплекс анионного типа, поэтому название заканчивается суффиксом -ат . Степень окисления центрального атома указывают римскими цифрами в скобках гексационоферрат (III) калия,
[Cu+2(NH3)6]+2SO4 комплекс катионного типа сульфат гексааммин меди (II),
[Co+3F3(H2O)3]o нейтральный комплекс (неэлектролит) центральный ион называют без указания его степени окисления триакватрифторокобальт, так как фтор в соединениях всегда F.
4.12. Равновесие в растворах комплексных соединений
При диссоциации в растворах и многих химических реакциях комплекс сохраняется:
[Ni(NH3)4]SO4 [Ni(NH3)4]2+ + SO42,
[Ni(NH3)4]SO4 + BaCl2 = [Ni(NH3)4]Cl2 + BaSO4 .
Обычно комплексные соединения в растворах диссоциируют на внешнюю и внутреннюю координационные сферы практически полностью по типу сильных электролитов (первичная диссоциация). Комплексные соединения обладают различной прочностью внутренней координационной сферы. Наряду с соединениями, внутренняя сфера которых отличается значительной прочностью и для которых диссоциация ничтожно мала, существуют соединения с крайне непрочной внутренней сферой. Растворы этих соединений практически не содержат комплексных ионов, так как они полностью диссоциируют на свои составные части (двойные соли). Диссоциация внутренней координационной сферы носит название вторичной, является обратимым процессом и проходит по типу слабых электролитов. Момент наступления равновесия характеризуется константой равновесия, которая в случае комплексного иона носит название константы нестойкости (КН).
Пример 8. Напишите процессы диссоциации и выражение для константы нестойкости комплексной соли К4[Fe(CN)6].
Решение. Если комплексная соль гексацианоферрат (III) калия, являясь сильным электролитом, в водном растворе практически полностью диссоциирует на ионы внешней и внутренней сфер:
K4[Fe(CN)6] = 4K+ + [Fe(CN)6]4 ,
то комплексный ион диссоциирует в незначительной степени на составляющие его частицы:
[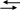 Fe(CN)6]4 Fe2+ + 6CN .
Fe(CN)6]4 Fe2+ + 6CN .
Константа равновесия этой реакции в данном случае является константой нестойкости (КН) комплекса:
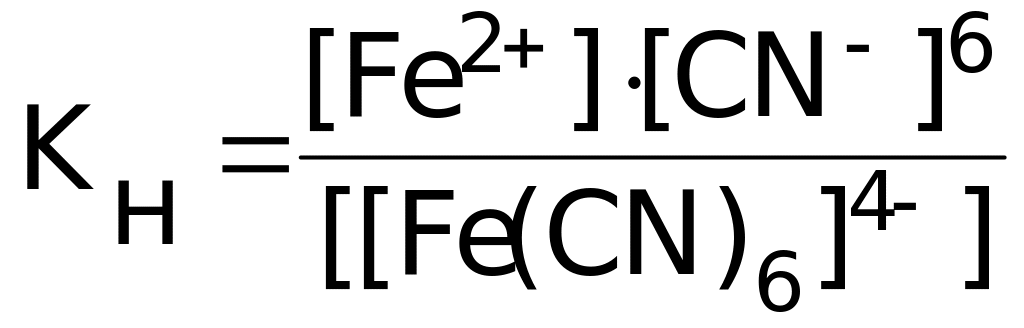 .
.
Значения констант нестойкости различных комплексных ионов колеблются в широких пределах и могут служить мерой устойчивости комплекса.
Чем меньше значение Кн, тем более прочен данный комплекс.
Сравним, например, константы нестойкости и устойчивость ряда комплексов серебра [Ag(NO2)2], [Ag(NH3)2]+, [Ag(S2O3)]3 и [Ag(CN)2]. Для них Кн: 1,3·103 ; 5,7·108; 2,5·1014 и 8,0·1021, соответственно. Следовательно, можно прийти к выводу, что наименее устойчивым из этих ионов является первый, а наиболее устойчивым последний. Очевидно также, что при одной и той же молярной концентрации комплексного соединения концентрация ионов Аg+ будет больше в растворе первого соединения и меньше в растворе последнего.
4.13. Химическая связь в комплексных соединениях
Для объяснения образования и свойств комплексных соединений в настоящее время применяется теория валентных связей (ВС), теория кристаллического поля (ТКП) и теория молекулярных орбиталей (МО). Мы ограничимся методом ВС.
Теория ВС для комплексных соединений. Пространственная структура комплексных частиц может быть объяснена с позиций метода (ВС). Согласно этому методу связь между центральным атомом и лигандами образуется за счет донорно-акцепторного взаимодействия: лиганд донор, а центральный атом акцептор электронной пары. При этом ковалентная -связь образуется в результате перекрывания вакантной орбитали центрального атома или иона комплексообразователя с заполненными, т.е. содержащими неподеленные пары электронов, орбиталями лигандов. Максимально возможное число -связей определяет координационное число комплексообразователя. Поскольку при одинаковых лигандах образующиеся -связи равноценны, то образование комплексной частицы сопровождается гибридизацией акцепторных орбиталей комплексообразователя. Критерием для определения типа гибридизации могут служить опытные данные о магнитных свойствах образующихся комплексов.
Пример 9. Определить пространственную структуру и устойчивость комплексных ионов: а) парамагнитного [CoF6]3 и б) диамагнитного [Co(NH3)6]3+.
Решение. а) [CoF6]3 координационное число Co равно 6, степень окисления () Co = +3; электронное строение атома Co …3d74s2, иона Co3+ ....3d64s04p04d0; для F ..2s22p5, для иона F .. 2s22p6.

(Крестом обозначена неподеленная пара электронов от лиганда иона F).
Из показанной схемы ВС следуют выводы:
Co3+ акцептор 6-ти электронных пар;
АО Co3+ гибридизованы, тип гибридизации sp3d2 – октаэдричес-
кий;
комплекс имеет 4 неспаренных электрона, он парамагнитен;
комплекс внешнеорбитальный, т.к. в образовании донорно-акцепторных связей принимают участие внешние 4d-АО (3d-АО внутренние орбитали). Использование внешних d-орбиталей требует затраты энергии, поэтому комплекс неустойчив.
б) [Co(NH3)6]3+ координационное число Co равно 6, степень окисления
()Сo = +3. Электронное строение NH3 таково, что каждая молекула имеет неподеленную электронную пару, за счет которой эта молекула будет донором. Так как комплекс диамагнитен, то все шесть d-электронов спарены, поэтому схему ВС для этого комплекса можно изобразить так:
![]()
Из этой диаграммы следуют выводы:
Co3+ акцептор 6-ти электронных пар;
АО (Co3+) гибридизованы по типу d2sp3 они образуют октаэдр;
комплекс диамагнитен, все электроны спарены;
комплекс внутриорбитальный, так как акцепторами являются внутренние 3d-атомные орбитали, он устойчивее, чем внешнеорбитальный.
Из приведенных примеров видны следующие особенности применения теории ВС к комплексам:
Для построения схемы ВС используются атомы и ионы с зарядом, соответствующим степени окисления.
Для образования химической связи не используются электроны центрального атома; используются лишь его пустые АО и электронные пары лигандов. (Однако возможен дополнительный учет дативного взаимодействия между парами d-электронов металла и пустыми (акцепторными) орбиталями лигандов).
Вопрос о расположении электронов на d-АО (и вопрос о магнитных свойствах) в теории ВС не решается, используются экспериментальные данные или данные других теорий (например, теория кристаллического поля ТКП).
В методе ВС вопросы геометрии комплекса, гибридизации АО, устойчивости решаются по схемам ВС.














Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!
Полезное для учителя

Реализация образовательных программ осуществляется с применением исключительно электронного обучения и ДОТ


